
L’uso dell’intelligenza artificiale (IA) nei dispositivi medici è in rapida crescita, offrendo nuove opportunità per migliorare la diagnosi e la cura dei pazienti.
Grazie all’IA, i dispositivi medici possono analizzare grandi quantità di dati in modo rapido ed efficiente, identificando pattern e anomalie che potrebbero sfuggire all’occhio umano.
Questo porta a diagnosi più accurate e tempestive, nonché a piani di trattamento personalizzati basati sulle esigenze specifiche di ciascun paziente.
Tuttavia, l’innovazione tecnologica porta con sé anche nuove sfide regolatorie.
La Food and Drug Administration (FDA) sta lavorando per definire un quadro normativo che garantisca la sicurezza e l’efficacia di questi dispositivi innovativi durante il loro intero ciclo di vita.
La regolazione dei dispositivi medici basati sull’IA è complessa, poiché questi dispositivi possono evolversi e migliorare continuamente attraverso l’apprendimento automatico.
Viene adottato un approccio proattivo, collaborando con sviluppatori di dispositivi, esperti di IA e altre parti interessate per creare linee guida che bilancino l’innovazione con la protezione dei pazienti.
Un aspetto cruciale di questo processo è la trasparenza: i produttori devono dimostrare chiaramente come i loro algoritmi funzionano e come vengono aggiornati. In questo modo, la FDA può valutare i rischi associati a ogni dispositivo e assicurarsi che siano gestiti adeguatamente.
Inoltre, sta esplorando l’uso di un quadro regolatorio basato su un ciclo di vita continuo, che permette ai dispositivi di essere monitorati e aggiornati costantemente una volta immessi sul mercato.
Questo approccio consente di affrontare rapidamente eventuali problemi di sicurezza e di incorporare miglioramenti tecnologici senza dover passare attraverso lunghi processi di riapprovazione.
La regolazione dei dispositivi medici basati sull’IA è ancora in evoluzione, ma la FDA è determinata a garantire che questi strumenti rivoluzionari possano essere utilizzati in modo sicuro ed efficace.
Con un quadro normativo robusto, l’IA ha il potenziale di trasformare radicalmente la sanità, migliorando i risultati per i pazienti e rendendo le cure più accessibili e personalizzate.
Principi di buona pratica di machine learning (GMLP)

La FDA, in collaborazione con Health Canada e MHRA del Regno Unito, ha recentemente pubblicato un documento contenente 10 principi guida per promuovere lo sviluppo delle Good Machine Learning Practice (GMLP) per i dispositivi medici basati su intelligenza artificiale e machine learning.
Questi principi mirano a gettare le basi per la creazione di dispositivi sicuri, efficaci e di alta qualità.Uno dei principi chiave è l’utilizzo di competenze multidisciplinari durante l’intero ciclo di vita del prodotto.
Principi Guida
- L’Esperienza Multidisciplinare è Sfruttata Durante l’Intero Ciclo di Vita del Prodotto: Una comprensione approfondita dell’integrazione prevista di un modello nel flusso di lavoro clinico, dei benefici desiderati e dei rischi associati per i pazienti può aiutare a garantire che i dispositivi medici abilitati all’IA siano sicuri ed efficaci e rispondano a esigenze clinicamente significative durante l’intero ciclo di vita del dispositivo.
- Sono Implementate Buone Pratiche di Ingegneria del Software e di Sicurezza: Il design del modello è implementato con attenzione ai “fondamentali”: buone pratiche di ingegneria del software, garanzia della qualità dei dati, gestione dei dati e pratiche robuste di sicurezza informatica. Queste pratiche includono una gestione metodica dei rischi e un processo di progettazione che possono catturare e comunicare adeguatamente le decisioni e le motivazioni relative alla progettazione, all’implementazione e alla gestione dei rischi, nonché garantire l’autenticità e l’integrità dei dati.
- I Partecipanti agli Studi Clinici e i Set di Dati Sono Rappresentativi della Popolazione di Pazienti Intesa: I protocolli di raccolta dei dati dovrebbero garantire che le caratteristiche rilevanti della popolazione di pazienti intesa (ad esempio, in termini di età, genere, sesso, razza ed etnia), l’uso e gli input di misurazione siano sufficientemente rappresentati in un campione di dimensioni adeguate nello studio clinico e nei set di dati di addestramento e test, in modo che i risultati possano essere ragionevolmente generalizzati alla popolazione di interesse. Questo è importante per gestire qualsiasi bias, promuovere prestazioni appropriate e generalizzabili nella popolazione di pazienti intesa, valutare l’usabilità e identificare le circostanze in cui il modello può avere prestazioni inferiori.
- I Set di Dati di Addestramento Sono Indipendenti dai Set di Test: I set di dati di addestramento e test sono selezionati e mantenuti in modo da essere opportunamente indipendenti l’uno dall’altro. Tutte le potenziali fonti di dipendenza, inclusi i fattori relativi ai pazienti, all’acquisizione dei dati e ai siti, sono considerate e affrontate per garantire l’indipendenza.
- I Set di Dati di Riferimento Selezionati Sono Basati sui Migliori Metodi Disponibili: I metodi accettati e migliori disponibili per lo sviluppo di un set di dati di riferimento (cioè uno standard di riferimento) garantiscono che vengano raccolti dati clinicamente rilevanti e ben caratterizzati e che le limitazioni del riferimento siano comprese. Se disponibili, vengono utilizzati set di dati di riferimento accettati nello sviluppo e nel test del modello per promuovere e dimostrare la robustezza e la generalizzabilità del modello nella popolazione di pazienti intesa.
- Il Design del Modello è Adattato ai Dati Disponibili e Riflette l’Uso Previsto del Dispositivo: Il design del modello è adatto ai dati disponibili e supporta la mitigazione attiva dei rischi noti, come l’overfitting, il degrado delle prestazioni e i rischi di sicurezza. I benefici clinici e i rischi relativi al prodotto sono ben compresi, utilizzati per derivare obiettivi di prestazione clinicamente significativi per i test e supportano che il prodotto possa raggiungere in modo sicuro ed efficace il suo uso previsto. Le considerazioni includono l’impatto delle prestazioni sia globali che locali e l’incertezza/variabilità negli input del dispositivo, negli output, nelle popolazioni di pazienti intese e nelle condizioni d’uso cliniche.
- L’Attenzione è Focalizzata sulle Prestazioni del Team Umano-IA: Quando il modello prevede un “umano nel loop”, vengono affrontate le considerazioni sui fattori umani e l’interpretabilità umana degli output del modello con enfasi sulle prestazioni del team Umano-IA, piuttosto che solo sulle prestazioni del modello in isolamento.
- I Test Dimostrano le Prestazioni del Dispositivo Durante Condizioni Clinicamente Rilevanti: Vengono sviluppati e eseguiti piani di test statisticamente solidi per generare informazioni sulle prestazioni del dispositivo clinicamente rilevanti indipendentemente dal set di dati di addestramento. Le considerazioni includono la popolazione di pazienti intesa, i sottogruppi importanti, l’ambiente clinico e l’uso da parte del team Umano-IA, gli input di misurazione e i potenziali fattori di confusione.
- Gli Utenti Sono Forniti di Informazioni Chiare ed Essenziali: Agli utenti viene fornito un accesso pronto a informazioni chiare e contestualmente rilevanti, appropriate per il pubblico previsto (come i fornitori di assistenza sanitaria o i pazienti) includendo: l’uso previsto del prodotto e le indicazioni per l’uso, le prestazioni del modello per i sottogruppi appropriati, le caratteristiche dei dati utilizzati per addestrare e testare il modello, gli input accettabili, le limitazioni conosciute, l’interpretazione dell’interfaccia utente e l’integrazione del modello nel flusso di lavoro clinico. Gli utenti sono anche informati delle modifiche e degli aggiornamenti del dispositivo derivanti dal monitoraggio delle prestazioni nel mondo reale, della base per il processo decisionale quando disponibile e di un mezzo per comunicare preoccupazioni sul prodotto allo sviluppatore.
- I Modelli Distribuiti Sono Monitorati per le Prestazioni e i Rischi di Riaddestramento Sono Gestiti: I modelli distribuiti hanno la capacità di essere monitorati nell’uso “reale” con un focus sul mantenimento o miglioramento della sicurezza e delle prestazioni. Inoltre, quando i modelli vengono periodicamente o continuamente addestrati dopo la distribuzione, ci sono controlli appropriati in atto per gestire i rischi di overfitting, bias non intenzionali o degrado del modello (ad esempio, drift del set di dati) che possono influenzare la sicurezza e le prestazioni del modello mentre viene utilizzato dal team Umano-IA.
Questi principi guida stabiliti dalla FDA, Health Canada e MHRA sono essenziali per garantire che l’IA nei dispositivi medici sia utilizzata in modo sicuro ed efficace. Promuovendo la collaborazione, l’innovazione e l’adozione di buone pratiche, questi principi aiutano a creare un ambiente regolatorio solido che protegge la salute pubblica e favorisce l’innovazione nel settore sanitario.
I principi sottolineano inoltre l’importanza di utilizzare set di dati rappresentativi della popolazione di pazienti designati durante lo sviluppo e il test dei modelli di machine learning, al fine di limitare rischi di bias e pregiudizi.
Viene anche sancito il principio dell’approccio “human-in-the-loop“, che pone la conoscenza e l’interpretabilità umana al centro dei processi di apprendimento automatico.
Questo approccio prevede l’integrazione continua dell’intervento umano nei processi decisionali dell’IA. In altre parole, mentre l’IA può elaborare e analizzare dati, le decisioni finali e le azioni critiche vengono sempre supervisionate o validate da operatori umani. Questo è particolarmente importante nel contesto medico, dove la precisione e la sicurezza sono essenziali.
Il “human-in-the-loop” consente di mitigare i rischi associati a errori algoritmici e di garantire che le decisioni prese dall’IA siano in linea con le migliori pratiche cliniche e le esigenze individuali dei pazienti. Inoltre, permette agli operatori sanitari di fornire un feedback continuo che può essere utilizzato per migliorare ulteriormente gli algoritmi di IA.
La FDA ha adottato un approccio proattivo, collaborando con sviluppatori di dispositivi, esperti di IA e altre parti interessate per creare linee guida che bilancino l’innovazione con la protezione dei pazienti.
Un aspetto cruciale di questo processo è la trasparenza: i produttori devono dimostrare chiaramente come i loro algoritmi funzionano e come vengono aggiornati. In questo modo, la FDA può valutare i rischi associati a ogni dispositivo e assicurarsi che siano gestiti adeguatamente.
Impatto della Normativa MDR sui Dispositivi Medici AI-based

L’ingresso dell’intelligenza artificiale (AI) self-learning nei dispositivi medici impone una rilettura delle norme del Regolamento sui Dispositivi Medici (MDR) e delle prescrizioni regolatorie di settore.
Al momento non esistono regolamenti o norme armonizzate che disciplinano specificatamente l’uso dell’AI nei dispositivi medici, ma questi devono comunque rispettare i requisiti regolatori stabilità dal Regolamento MDR (UE) 2017/745.
Definizione e Applicabilità
Il Regolamento (UE) 2017/745, noto come Medical Device Regulation (MDR), è una normativa dell’Unione Europea che disciplina i dispositivi medici. Pubblicato nella Gazzetta ufficiale dell’Unione europea il 5 maggio 2017, è entrato in vigore il 26 maggio 2017, con piena applicabilità dal 26 maggio 2021
Il MDR ha ampliato la definizione di dispositivo medico, aumentando il numero di prodotti soggetti alla nuova normativa.
Il Regolamento MDR si applica sia ai software che hanno l’obiettivo di diagnosticare, prevenire, monitorare, trattare o attenuare una malattia, sia a quelli che forniscono informazioni destinate a informare un operatore sanitario nel prendere la decisione diagnostica o terapeutica finale.I fabbricanti di software per dispositivi devono soddisfare i requisiti generali di sicurezza e prestazioni, verificando che il software sia in grado di generare l’output previsto in modo accurato, affidabile e preciso.
Il MDR si applica a tutti i dispositivi medici immessi sul mercato o messi in servizio nell’UE.
Include una vasta gamma di prodotti, dai dispositivi di classe I (a basso rischio) ai dispositivi di classe III (ad alto rischio), e copre anche i software destinati a scopi medici (Software as Medical Device – SAMD)
Il MDR prevede anche specifiche norme relative alla qualificazione del software come dispositivo medico.
Obiettivi del MDR
Il MDR mira a garantire:
- Un elevato livello di protezione della salute e sicurezza dei pazienti.
- Il buon funzionamento del mercato interno dell’UE.
- Maggiore trasparenza e tracciabilità dei dispositivi medici.
Principali Novità e Ambito di Applicazione
- Ampliamento della Definizione di Dispositivo Medico: Il MDR ha esteso la definizione di dispositivo medico, includendo una gamma più ampia di prodotti.
- Requisiti di Sicurezza e Prestazione: Tutti i dispositivi devono rispettare requisiti dettagliati per garantire un elevato livello di qualità e sicurezza.
- Responsabilità degli Operatori Economici: Aumento delle responsabilità per produttori, importatori e distributori lungo tutta la catena di fornitura.
- Valutazione della Conformità: Procedure più rigorose per la valutazione della conformità, comprese indagini cliniche e valutazioni del rischio.
- Sorveglianza del Mercato: Maggiore enfasi sul monitoraggio continuo delle prestazioni dei dispositivi durante il loro ciclo di vita.
Modifiche Normative
Il MDR ha introdotto modifiche significative in vari aspetti:
1. Immissione sul mercato
Il MDR ha rafforzato i requisiti per l’immissione sul mercato dei dispositivi medici. Le aziende devono fornire prove più solide della sicurezza e delle prestazioni dei loro prodotti prima di poterli commercializzare. Questo include una documentazione tecnica più dettagliata e rigorosi requisiti di conformità clinica [1].
2. Supervisione degli organismi notificati
Il regolamento ha introdotto una supervisione più rigorosa degli organismi notificati, ovvero le entità responsabili della valutazione della conformità dei dispositivi medici. Gli organismi notificati devono ora soddisfare criteri più stringenti per ottenere e mantenere la loro designazione, e sono soggetti a una sorveglianza continua da parte delle autorità competenti [2].
3. Procedure di valutazione della conformità
Le procedure di valutazione della conformità sono state ampliate e rese più rigorose. Le aziende devono sottoporre i loro dispositivi a valutazioni più dettagliate, che includono prove cliniche e una continua sorveglianza post-commercializzazione. Questo aiuta a garantire che i dispositivi rimangano sicuri ed efficaci durante tutto il loro ciclo di vita [3].
4. Indagini e valutazioni cliniche
Il MDR richiede che tutte le indagini e valutazioni cliniche siano eseguite in conformità con standard elevati. Le prove cliniche devono essere condotte in modo trasparente e devono fornire dati solidi sulla sicurezza e sulle prestazioni dei dispositivi medici. Questo è essenziale per ottenere l’approvazione per l’immissione sul mercato [4].
5. Valutazione del rischio
La valutazione del rischio è stata rafforzata per garantire che tutti i potenziali rischi associati ai dispositivi medici siano identificati, valutati e mitigati. Le aziende devono implementare un sistema di gestione del rischio che copra tutte le fasi del ciclo di vita del dispositivo, dalla progettazione alla post-commercializzazione [1].
6. Sorveglianza del mercato
Il MDR ha introdotto misure più rigorose per la sorveglianza del mercato, con l’obiettivo di monitorare continuamente i dispositivi medici una volta che sono stati immessi sul mercato. Le autorità competenti hanno maggiori poteri per intervenire in caso di problemi di sicurezza e per garantire che i dispositivi non conformi siano rapidamente ritirati dal mercato [2].
Le modifiche introdotte dal MDR rappresentano un significativo passo avanti nella regolamentazione dei dispositivi medici, con l’obiettivo di migliorare la sicurezza dei pazienti e garantire che i dispositivi medici siano efficaci e conformi agli standard più elevati. Queste modifiche richiedono un impegno maggiore da parte delle aziende, ma sono essenziali per mantenere la fiducia nel sistema di regolamentazione dei dispositivi medici nell’Unione Europea.
Responsabilità degli Operatori Economici
Il MDR ha introdotto nuovi obblighi per tutti gli operatori economici coinvolti nella catena di fornitura, inclusi i fabbricanti, i rappresentanti autorizzati, gli importatori e i distributori.
Questi obblighi mirano a garantire che ogni dispositivo medico immesso sul mercato europeo sia conforme ai requisiti di sicurezza e prestazione stabiliti dal regolamento. Gli operatori economici devono assicurarsi che i dispositivi siano correttamente etichettati, accompagnati dalla documentazione richiesta e che siano stati sottoposti alle necessarie valutazioni di conformità
Obblighi di Controllo
Il regolamento impone agli operatori economici di mantenere un sistema di tracciabilità efficace per tutti i dispositivi medici. Questo include la registrazione e la conservazione delle informazioni relative ai dispositivi, come i numeri di lotto o di serie, e la possibilità di rintracciare i dispositivi lungo tutta la catena di fornitura. Inoltre, gli operatori economici devono collaborare con le autorità competenti per facilitare le attività di vigilanza sul mercato e di sorveglianza post-commercializzazione [2].
Obblighi per gli Utilizzatori e le Strutture Sanitarie
Anche gli utilizzatori, comprese le strutture sanitarie, hanno nuovi obblighi sotto il MDR. Devono rispettare istruzioni d’uso più dettagliate e precise fornite dai fabbricanti. Questo include l’adozione di misure per garantire che i dispositivi siano utilizzati correttamente e in conformità con le istruzioni del fabbricante, e la segnalazione di qualsiasi incidente o problema di sicurezza alle autorità competenti e al fabbricante stesso [3].
Benefici per la Sicurezza dei Pazienti
Questi cambiamenti sono stati introdotti per migliorare la sicurezza dei pazienti e aumentare la trasparenza e la tracciabilità dei dispositivi medici. Garantendo che tutti gli operatori economici e utilizzatori rispettino rigorosi obblighi di controllo e tracciabilità, il MDR mira a ridurre il rischio di dispositivi non conformi o pericolosi sul mercato e a migliorare la capacità di risposta in caso di problemi di sicurezza
Ruolo del Ministero della Salute
Il Ministero della Salute italiano è impegnato in diverse attività per garantire la conformità dei dispositivi medici al nuovo Regolamento (UE) 2017/745.
Ecco un’analisi dettagliata delle principali azioni intraprese:
Supporto agli Organismi Notificati Italiani
Il Ministero della Salute fornisce supporto agli organismi notificati italiani per aiutarli a conformarsi ai nuovi requisiti del MDR.
Questo include assistenza tecnica e operativa per garantire che gli organismi notificati possano svolgere adeguatamente le loro funzioni di valutazione della conformità dei dispositivi medici.
Partecipazione ai Gruppi di Lavoro e Task Force dell’MDCG
Il Ministero partecipa attivamente ai gruppi di lavoro e alle task force del Medical Device Coordination Group (MDCG), un organismo istituito dalla Commissione Europea per facilitare l’implementazione armonizzata del MDR.
La partecipazione a questi gruppi consente al Ministero di contribuire allo sviluppo di linee guida e di condividere le migliori pratiche con altri Stati membri
Integrazione dei Sistemi di Registrazione Nazionali ed Europei
Un’altra importante attività del Ministero è l’integrazione dei sistemi di registrazione nazionali con quelli europei.
Questo include la connessione al sistema Eudamed, una banca dati europea per i dispositivi medici, che migliora la trasparenza e la tracciabilità dei dispositivi medici in tutta l’UE. L’integrazione facilita la condivisione delle informazioni e il monitoraggio dei dispositivi medici
Adeguamento della Normativa Nazionale al Regolamento (UE) 2017/745
Il Ministero della Salute sta lavorando all’adeguamento della normativa nazionale per allinearla ai requisiti del MDR.
Questo processo include la revisione e l’aggiornamento delle leggi e dei regolamenti esistenti per garantire che siano conformi alle nuove disposizioni europee. L’adeguamento normativo è essenziale per assicurare una corretta applicazione del MDR a livello nazionale
Indagini Cliniche e Vigilanza
Il Ministero ha emesso circolari per fornire indicazioni operative sulle indagini cliniche e sulla vigilanza dei dispositivi medici.
Queste circolari offrono linee guida dettagliate su come condurre indagini cliniche in conformità con il MDR e su come monitorare la sicurezza e le prestazioni dei dispositivi medici una volta immessi sul mercato. In attesa dell’adeguamento completo della normativa nazionale e della piena operatività del sistema Eudamed, queste indicazioni operative sono cruciali per garantire la conformità e la sicurezza dei dispositivi medici
Le attività del Ministero della Salute sono fondamentali per garantire che i dispositivi medici in Italia siano conformi al nuovo Regolamento (UE) 2017/745. Attraverso il supporto agli organismi notificati, la partecipazione ai gruppi di lavoro dell’MDCG, l’integrazione dei sistemi di registrazione, l’adeguamento normativo e la vigilanza, il Ministero sta lavorando per migliorare la sicurezza e l’efficacia dei dispositivi medici nel paese.
Implementazione Nazionale
In Italia, l’adeguamento alla normativa MDR è stato realizzato con il Decreto Legislativo n. 137 del 5 agosto 2022, che armonizza la legislazione nazionale con il nuovo quadro europeo.
Il Decreto Legislativo n. 137 del 5 agosto 2022 è stato emanato per adeguare la normativa nazionale italiana alle disposizioni del Regolamento (UE) 2017/745 relativo ai dispositivi medici.
Questo decreto è stato pubblicato nella Gazzetta Ufficiale Serie Generale n. 214 del 13 settembre 2022 e ha lo scopo di garantire che tutti i soggetti coinvolti, come organismi notificati, operatori economici e operatori sanitari, rispettino le nuove normative europee
Principali Disposizioni del Decreto Legislativo n. 137/2022
Il Decreto Legislativo n. 137 del 5 agosto 2022 adegua la normativa nazionale alle disposizioni del Regolamento (UE) 2017/745 sui dispositivi medici. Ecco un riassunto delle principali disposizioni:
Marcatura CE
I dispositivi medici su misura non recano la marcatura CE ma devono comunque soddisfare gli obblighi previsti dal Regolamento (UE) 2017/745. Questo significa che, sebbene tali dispositivi non abbiano il marchio CE, devono comunque essere conformi ai requisiti di sicurezza e prestazione stabiliti dal regolamento. La mancata apposizione della marcatura CE è giustificata dal fatto che questi dispositivi sono realizzati specificamente per un singolo paziente, in base a una prescrizione medica, e quindi non rientrano nelle stesse categorie di conformità dei dispositivi prodotti in serie
Procedura di Conformità
I fabbricanti di dispositivi medici devono seguire una procedura di conformità rigorosa, come delineato nell’allegato XIII del Regolamento (UE) 2017/745. Questa procedura include diversi elementi chiave:
- Rispetto dei Requisiti Generali di Sicurezza e Prestazione: I fabbricanti devono garantire che i loro dispositivi soddisfino i requisiti generali di sicurezza e prestazione stabiliti nell’allegato I del regolamento. Questo implica una valutazione completa dei rischi associati e l’adozione di misure appropriate per mitigarli
- Motivazione per Eventuali Requisiti Non Rispettati: Se alcuni requisiti non possono essere rispettati, i fabbricanti devono fornire una motivazione dettagliata e documentata. Questo aiuta a garantire trasparenza e responsabilità nel processo di conformità [3].
- Sistema di Gestione della Qualità: I fabbricanti devono implementare un sistema di gestione della qualità proporzionale alla classe di rischio del dispositivo. Questo sistema deve coprire tutti gli aspetti della progettazione, produzione e controllo post-commercializzazione del dispositivo
- Documentazione Relativa alla Progettazione, Fabbricazione e Prestazioni del Dispositivo: I fabbricanti devono mantenere una documentazione completa e dettagliata che dimostri la conformità del dispositivo ai requisiti normativi. Questa documentazione deve includere tutte le fasi del ciclo di vita del prodotto, dalla progettazione alla produzione e alle prestazioni cliniche [1].
- Implementazione di un Sistema di Sorveglianza Post-Commercializzazione: I fabbricanti devono implementare un sistema di sorveglianza post-commercializzazione per monitorare continuamente la sicurezza e le prestazioni del dispositivo una volta immesso sul mercato. Questo sistema deve includere la raccolta e l’analisi dei dati relativi all’uso del dispositivo e la segnalazione di eventuali problemi [2].
- Segnalazione degli Incidenti Gravi e delle Azioni Correttive di Sicurezza: È obbligatorio per i fabbricanti segnalare tempestivamente qualsiasi incidente grave e le azioni correttive di sicurezza alle autorità competenti. Questo è cruciale per garantire una risposta rapida ed efficace a qualsiasi problema di sicurezza che possa emergere
- Nomina di una Persona Responsabile del Rispetto della Normativa: I fabbricanti devono nominare una persona responsabile del rispetto della normativa, che abbia le competenze necessarie per garantire che il dispositivo sia conforme ai requisiti del regolamento. Questa figura è essenziale per assicurare la conformità continua e la gestione dei rischi associati ai dispositivi medici [4].
Le disposizioni del Decreto Legislativo n. 137/2022 sono fondamentali per garantire che i dispositivi medici siano sicuri ed efficaci, rispettando i rigorosi requisiti stabiliti dal Regolamento (UE) 2017/745.
Attraverso la marcatura CE, la procedura di conformità, la sorveglianza post-commercializzazione e la nomina di una persona responsabile, il decreto mira a migliorare la sicurezza dei pazienti e a garantire la qualità dei dispositivi medici sul mercato.
Il Decreto Legislativo n. 137/2022 e il Regolamento (UE) 2017/745 stabiliscono specifici obblighi e responsabilità per i fabbricanti di dispositivi medici su misura. Ecco una spiegazione dettagliata delle principali disposizioni:
Valutazione della Conformità
Per i dispositivi su misura di classe III impiantabili, è richiesto l’intervento di un organismo notificato per la valutazione della conformità. Questo significa che i dispositivi di questa categoria devono essere sottoposti a una rigorosa valutazione da parte di un ente indipendente, che verifica che il dispositivo soddisfi tutti i requisiti di sicurezza e prestazione stabiliti dal regolamento. Questo processo è essenziale per garantire che i dispositivi impiantabili su misura siano sicuri ed efficaci per l’uso clinico [1].
Iscrizione nell’Elenco del Ministero della Salute
I fabbricanti di dispositivi medici su misura devono iscriversi nell’elenco del Ministero della Salute. Questo obbligo è in continuità con quanto previsto dal Decreto Legislativo 46/97. L’iscrizione nell’elenco è necessaria per garantire che tutti i fabbricanti operino in conformità con le normative vigenti e per facilitare la sorveglianza e il controllo da parte delle autorità competenti. Questo elenco funge da registro ufficiale dei fabbricanti autorizzati, contribuendo a mantenere elevati standard di qualità e sicurezza nel settore dei dispositivi medici [2].
Le disposizioni relative agli obblighi e alle responsabilità dei fabbricanti di dispositivi medici su misura sono fondamentali per garantire la conformità ai requisiti normativi e la sicurezza dei pazienti. Attraverso la valutazione della conformità da parte di organismi notificati e l’iscrizione nell’elenco del Ministero della Salute, il decreto legislativo mira a rafforzare la regolamentazione e il controllo dei dispositivi medici su misura, assicurando che siano sicuri, efficaci e conformi agli standard più elevati [3], [4].
Il Decreto Legislativo n. 137/2022, in conformità con il Regolamento (UE) 2017/745, stabilisce criteri rigorosi per la pubblicità dei dispositivi medici, mirati a garantire trasparenza e veridicità. Ecco una spiegazione dettagliata delle principali disposizioni:
Veridicità delle Informazioni
Tutte le informazioni pubblicitarie relative ai dispositivi medici devono essere accurate e supportate da evidenze scientifiche. È vietato fare affermazioni ingannevoli o non supportate da dati clinici verificabili. Le informazioni devono riflettere fedelmente l’efficacia e la sicurezza del dispositivo, evitando qualsiasi dichiarazione che possa indurre in errore i consumatori. Questo è essenziale per mantenere la fiducia del pubblico e per garantire che le decisioni dei consumatori siano basate su dati reali e affidabili [1].
Conformità alla Normativa
La pubblicità dei dispositivi medici deve essere conforme alle normative vigenti, incluse le specifiche disposizioni del Regolamento (UE) 2017/745. Questo include l’obbligo di non fare affermazioni che superino le indicazioni approvate per l’uso del dispositivo. Le aziende devono assicurarsi che tutte le comunicazioni pubblicitarie rispettino i limiti imposti dalle autorizzazioni e dalle certificazioni ottenute per i loro prodotti
Autorizzazione Preventiva
In alcuni casi, potrebbe essere richiesta un’autorizzazione preventiva da parte delle autorità competenti prima di avviare campagne pubblicitarie. Questo è particolarmente rilevante per i dispositivi medici di classe elevata o a rischio maggiore, dove la verifica preventiva delle informazioni pubblicitarie può essere necessaria per garantire che non vi siano affermazioni fuorvianti o non supportate. L’autorizzazione preventiva aiuta a prevenire la diffusione di informazioni potenzialmente dannose per la salute pubblica [3].
Le disposizioni del Decreto Legislativo n. 137/2022 relative alla pubblicità dei dispositivi medici sono fondamentali per garantire che le informazioni fornite ai consumatori siano accurate, trasparenti e conformi alle normative. Attraverso la veridicità delle informazioni, la conformità alla normativa e, in alcuni casi, l’autorizzazione preventiva, il decreto mira a proteggere i consumatori e a mantenere elevati standard di qualità e sicurezza nella comunicazione pubblicitaria dei dispositivi medici
Il Decreto Legislativo n. 137/2022 prevede diverse misure per garantire la sicurezza e la conformità dei dispositivi medici venduti tramite piattaforme digitali. Ecco una spiegazione dettagliata delle principali disposizioni:
Registrazione e Autorizzazione
I venditori online di dispositivi medici devono essere registrati e autorizzati dalle autorità competenti. Questo assicura che solo gli operatori che rispettano i requisiti normativi possano vendere dispositivi medici online. Inoltre, i venditori devono garantire che i dispositivi medici venduti siano conformi alle normative europee e nazionali, assicurando che i prodotti siano sicuri ed efficaci [1].
Informazioni Trasparenti
Le piattaforme di vendita online devono fornire informazioni chiare e trasparenti sui dispositivi medici. Questo include dettagli sulla conformità del dispositivo, istruzioni per l’uso, e informazioni sulla sicurezza. Le piattaforme devono anche fornire un contatto diretto per assistenza e ulteriori informazioni, facilitando così l’accesso degli utenti a supporto e chiarimenti [2].
Sorveglianza Post-Vendita
I venditori online sono tenuti a implementare sistemi di sorveglianza post-vendita per monitorare l’uso dei dispositivi medici. Questo sistema deve includere la raccolta e l’analisi dei dati relativi all’uso del dispositivo e la segnalazione di eventuali incidenti o problemi di sicurezza alle autorità competenti. La sorveglianza post-vendita è cruciale per identificare e risolvere rapidamente eventuali problemi che possano emergere dopo che il dispositivo è stato immesso sul mercato [3].
Divieti Specifici
È vietato vendere online dispositivi medici che non siano stati approvati o che non abbiano superato le procedure di valutazione della conformità. Inoltre, devono essere rispettate tutte le restrizioni relative alla pubblicità e alla promozione dei dispositivi medici. Questo assicura che solo i dispositivi che hanno dimostrato di essere sicuri ed efficaci possano essere venduti e promossi online, proteggendo così la salute dei consumatori [4].
Le disposizioni del Decreto Legislativo n. 137/2022 relative alla vendita online dei dispositivi medici sono state introdotte per garantire che i dispositivi venduti e pubblicizzati online siano sicuri, efficaci e conformi alle normative vigenti. Attraverso la registrazione e l’autorizzazione dei venditori, la trasparenza delle informazioni, la sorveglianza post-vendita e il rispetto dei divieti specifici, il decreto mira a proteggere la salute dei consumatori e a mantenere elevati standard di qualità nel mercato dei dispositivi medici [1], [2], [3], [4].
La compatibilità con Eudamed è un aspetto cruciale per garantire la trasparenza e la tracciabilità dei dispositivi medici nell’Unione Europea. Ecco un ampliamento del contenuto basato su alcune fonti:
Regolamento (UE) 2017/745
Questo regolamento stabilisce che tutti i dispositivi medici devono essere registrati nella banca dati europea Eudamed. Il sistema Eudamed consente di raccogliere e condividere informazioni sui dispositivi medici, migliorando la trasparenza e la sicurezza dei pazienti. La compatibilità con Eudamed richiede che le banche dati nazionali siano costantemente allineate e aggiornate in conformità con il Sistema unico di identificazione del dispositivo (UDI)
Decreto del Ministro della Salute del 2 agosto 2005
Questo decreto specifica le modalità di presentazione della documentazione necessaria per la notifica delle indagini cliniche sui dispositivi medici. È essenziale che i dati raccolti siano compatibili con Eudamed per garantire un’integrazione fluida e l’accesso alle informazioni a livello europeo. Questo allineamento è fondamentale per il monitoraggio continuo e la sicurezza dei dispositivi medici [2].
Decreti Legislativi per l’Attuazione del Regolamento (UE) 2017/745
Pubblicati sulla Gazzetta Ufficiale, questi decreti tengono conto della necessità di garantire la compatibilità tra le banche dati nazionali e Eudamed. Essi stabiliscono le norme per l’implementazione del Sistema unico di identificazione del dispositivo (UDI), che è essenziale per tracciare i dispositivi medici e garantire la loro sicurezza e conformità alle normative europee
Relazione Illustrativa sullo Schema di Decreto Legislativo
La relazione sottolinea l’importanza di determinare la sicurezza e la compatibilità dei dispositivi medici con i potenziali soggetti riceventi. Favorire la connessione dei sistemi nazionali con Eudamed è essenziale per migliorare la qualità e l’affidabilità dei dati sui dispositivi medici, permettendo una gestione più efficiente delle informazioni e una risposta rapida in caso di problemi di sicurezza [4].
La compatibilità con Eudamed rappresenta un elemento chiave per garantire che i dispositivi medici siano tracciabili e trasparenti in tutta l’Unione Europea. Attraverso l’allineamento delle banche dati nazionali, la presentazione corretta della documentazione e l’implementazione dell’UDI, si mira a migliorare la sicurezza e la qualità dei dispositivi medici disponibili sul mercato europeo [1], [2], [3], [4].
Il Decreto Legislativo n. 137/2022 mira a raggiungere diversi obiettivi chiave per migliorare la regolamentazione e la gestione dei dispositivi medici in Italia. Ecco una spiegazione dettagliata delle principali finalità del decreto:
Assicurare un’Armonizzazione Completa con le Normative Europee
Il decreto è stato sviluppato per garantire una piena armonizzazione con le normative europee, in particolare con il Regolamento (UE) 2017/745. Questo include l’adozione di tutte le disposizioni necessarie per assicurare che i dispositivi medici immessi sul mercato italiano siano conformi agli standard europei, migliorando così la coerenza e l’affidabilità dei dispositivi medici in tutta l’UE
Migliorare la Sicurezza e la Qualità dei Dispositivi Medici
Un obiettivo fondamentale del decreto è migliorare la sicurezza e la qualità dei dispositivi medici disponibili sul mercato. Questo viene realizzato attraverso l’implementazione di rigorose procedure di valutazione della conformità, la sorveglianza post-commercializzazione e la necessità di prove cliniche solide per dimostrare la sicurezza e l’efficacia dei dispositivi [2].
Facilitare la Tracciabilità e la Trasparenza nel Mercato dei Dispositivi Medici
Il decreto introduce misure per facilitare la tracciabilità e la trasparenza dei dispositivi medici. Questo include l’obbligo di registrazione dei dispositivi nella banca dati europea Eudamed, che consente di raccogliere e condividere informazioni sui dispositivi medici, migliorando la trasparenza e la sicurezza dei pazienti
Rafforzare le Procedure di Approvvigionamento e la Governance dei Dispositivi Medici
Il decreto mira a rafforzare le procedure di approvvigionamento e la governance dei dispositivi medici attraverso l’Health Technology Assessment (HTA) e l’Osservatorio dei prezzi di acquisto dei dispositivi. L’HTA è uno strumento essenziale per valutare l’efficacia e il valore dei dispositivi medici, mentre l’Osservatorio dei prezzi aiuta a monitorare e controllare i costi, garantendo che le risorse siano utilizzate in modo efficiente e che i pazienti abbiano accesso a dispositivi medici di alta qualità
Il Decreto Legislativo n. 137/2022 rappresenta un passo significativo verso il miglioramento della regolamentazione dei dispositivi medici in Italia. Attraverso l’armonizzazione con le normative europee, il miglioramento della sicurezza e della qualità, la facilitazione della tracciabilità e della trasparenza, e il rafforzamento delle procedure di approvvigionamento e governance, il decreto mira a proteggere la salute dei pazienti e a garantire l’efficacia e la sicurezza dei dispositivi medici disponibili sul mercato
Il Decreto Legislativo n. 137/2022 impone ai fabbricanti di dispositivi medici di rispettare nuovi requisiti di conformità, fondamentali per garantire la sicurezza e l’efficacia dei dispositivi medici. Di seguito una spiegazione dettagliata delle principali disposizioni:
Redazione di una Documentazione Tecnica Dettagliata
I fabbricanti devono redigere una documentazione tecnica dettagliata per ciascun dispositivo medico. Questa documentazione deve includere tutte le informazioni necessarie per dimostrare la conformità del dispositivo ai requisiti del Regolamento (UE) 2017/745. Tra gli elementi inclusi nella documentazione tecnica vi sono descrizioni dettagliate del dispositivo, i risultati delle valutazioni cliniche, i dati di progettazione e produzione, e le prove di conformità alle norme applicabili [1].
Sorveglianza Post-Commercializzazione
I fabbricanti sono obbligati a implementare un sistema di sorveglianza post-commercializzazione per monitorare continuamente la sicurezza e le prestazioni dei dispositivi medici una volta immessi sul mercato. Questo sistema deve includere la raccolta e l’analisi dei dati relativi all’uso del dispositivo e la segnalazione di eventuali incidenti o problemi di sicurezza alle autorità competenti. Inoltre, i fabbricanti devono redigere rapporti periodici di aggiornamento sulla sicurezza (PSUR) per i dispositivi di classe superiore, che devono essere sottoposti a revisione continua [2].
Coinvolgimento di Organismi Notificati (ON) per la Valutazione della Conformità
Per i dispositivi medici di classe superiore, è richiesto il coinvolgimento di Organismi Notificati (ON) per la valutazione della conformità. Gli Organismi Notificati sono enti indipendenti autorizzati a valutare se un dispositivo medico soddisfa i requisiti di sicurezza e prestazione stabiliti dal Regolamento (UE) 2017/745. La valutazione da parte di un ON è necessaria per garantire che i dispositivi di classe superiore siano sicuri ed efficaci prima di essere immessi sul mercato [3].
Gli obblighi per i fabbricanti di dispositivi medici stabiliti dal Decreto Legislativo n. 137/2022 sono fondamentali per garantire la conformità ai requisiti normativi e la sicurezza dei pazienti. Attraverso la redazione di una documentazione tecnica dettagliata, la sorveglianza post-commercializzazione e il coinvolgimento degli Organismi Notificati per la valutazione della conformità, il decreto mira a migliorare la sicurezza e la qualità dei dispositivi medici disponibili sul mercato
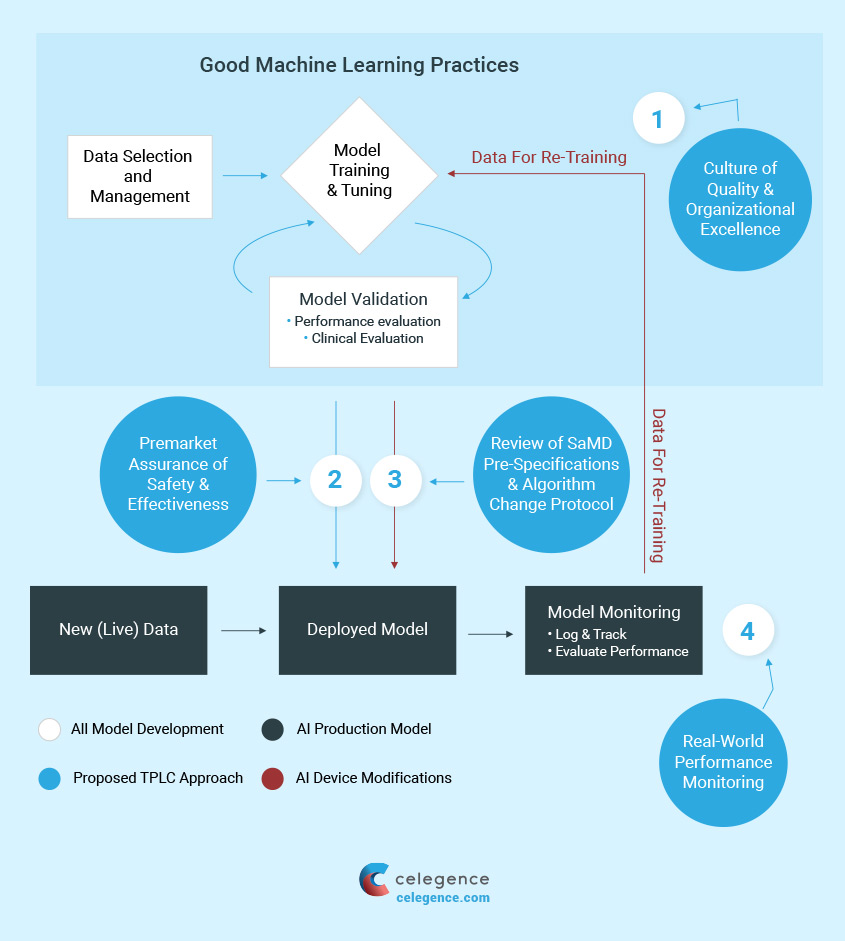
Potenziale rivoluzionario dell’IA in sanità
I processi complessi e dinamici coinvolti nello sviluppo, implementazione, utilizzo e manutenzione delle tecnologie di IA richiedono una gestione attenta durante l’intero ciclo di vita del prodotto medico.
In particolare, la gestione end-to-end delle applicazioni di IA è un processo iterativo che parte dall’ideazione e progettazione e procede attraverso l’acquisizione dei dati, la preparazione, lo sviluppo e la valutazione del modello, l’implementazione, il monitoraggio e manutenzione.
Questo approccio può aiutare ad affrontare le prestazioni continue del modello, la gestione del rischio e la conformità normativa dei sistemi di IA nelle applicazioni del mondo reale.
È importante sottolineare che la gestione dell’IA richiede un quadro normativo basato sul rischio, costruito su principi robusti, standard, migliori pratiche e strumenti all’avanguardia di scienza normativa che possono essere applicati a tutte le applicazioni di IA e adattati al prodotto medico pertinente .
CBER, CDER, CDRH e OCP
sono quattro centri e uffici all’interno della Food and Drug Administration (FDA) degli Stati Uniti, ciascuno con specifiche responsabilità nella regolamentazione di diversi tipi di prodotti medici.
CBER (Center for Biologics Evaluation and Research)
Il CBER è responsabile della regolamentazione dei prodotti biologici per uso umano, inclusi sangue, vaccini, allergenici, tessuti e terapie cellulari e geniche. La missione del CBER è proteggere e migliorare la salute pubblica attraverso la regolamentazione di questi prodotti, assicurando che siano sicuri, efficaci e disponibili per chi ne ha bisogno.
Il CBER valuta i dati scientifici e clinici presentati dai produttori per determinare se un prodotto biologico soddisfa gli standard di approvazione
CDER (Center for Drug Evaluation and Research)
Il CDER regola i farmaci da banco e su prescrizione, inclusi i farmaci biologici terapeutici e i farmaci generici. Il CDER assicura che i farmaci siano sicuri ed efficaci per migliorare la salute delle persone negli Stati Uniti.
Questo centro copre una vasta gamma di prodotti, dai medicinali ai prodotti come dentifrici al fluoro e shampoo antiforfora. Il CDER esamina le domande di nuovi farmaci, interagisce con l’industria farmaceutica e decide se i benefici di un farmaco superano i rischi noti
CDRH (Center for Devices and Radiological Health)
Il CDRH è responsabile della protezione e promozione della salute pubblica garantendo che i pazienti e i fornitori abbiano accesso tempestivo e continuo a dispositivi medici sicuri, efficaci e di alta qualità, nonché a prodotti che emettono radiazioni sicure.
Il CDRH facilita l’innovazione dei dispositivi medici avanzando la scienza regolatoria e fornendo percorsi regolatori prevedibili, coerenti, trasparenti ed efficienti
OCP (Office of Combination Products)
L’OCP assegna i prodotti combinati (che possono includere combinazioni di farmaci, dispositivi e prodotti biologici) ai centri di prodotti medici della FDA per la revisione, coordina le revisioni tempestive ed efficaci dei prodotti combinati e supervisiona le attività post-mercato per garantire la sicurezza e la qualità di questi prodotti.
L’OCP sviluppa linee guida, regolamenti e procedure operative standard per chiarire la regolamentazione dei prodotti combinati e facilita la risoluzione delle controversie riguardanti la tempestività delle revisioni pre-mercato
Questi centri e uffici lavorano insieme per garantire che i prodotti medici siano sicuri, efficaci e disponibili per il pubblico, ciascuno con un focus specifico su diverse categorie di prodotti.
Le quattro aree di interesse per CBER (Center for Biologics Evaluation and Research), CDER (Center for Drug Evaluation and Research), CDRH (Center for Devices and Radiological Health) e OCP (Office of Combination Products) riguardo allo sviluppo e all’uso dell’intelligenza artificiale (IA) nel ciclo di vita del prodotto medico sono fondamentali per garantire la sicurezza, l’efficacia e l’innovazione nel settore sanitario.
Ecco una spiegazione dettagliata di ciascuna area:
1. Promuovere la collaborazione per salvaguardare la salute pubblica
Questa area di interesse si concentra sull’importanza della collaborazione tra diversi enti regolatori, l’industria, il mondo accademico e altri stakeholder per garantire che l’uso dell’IA nei prodotti medici sia sicuro e non comprometta la salute pubblica.
La collaborazione può includere la condivisione di dati, esperienze e competenze per identificare e mitigare i potenziali rischi associati all’IA.
L’obiettivo è creare un ambiente in cui tutte le parti interessate lavorino insieme per proteggere i pazienti e migliorare la qualità dei prodotti medici.
2. Promuovere lo sviluppo di approcci normativi che supportano l’innovazione
Per supportare l’innovazione, è essenziale sviluppare approcci normativi flessibili e adattabili che possano tenere il passo con i rapidi progressi tecnologici nell’IA.
Questo include la creazione di linee guida e regolamenti che facilitino l’approvazione e l’adozione di nuove tecnologie IA nei prodotti medici, senza compromettere la sicurezza e l’efficacia.
In pratica, significa che le normative dovrebbero essere progettate in modo da non ostacolare l’innovazione, ma piuttosto da incoraggiarla, garantendo al contempo che i nuovi prodotti siano sicuri per i pazienti.
3. Promuovere lo sviluppo di standard, linee guida, migliori pratiche e strumenti per il ciclo di vita del prodotto medico
Questa area riguarda la definizione di standard e linee guida che possano essere utilizzati durante tutto il ciclo di vita del prodotto medico, dall’ideazione alla post-commercializzazione.
Lo sviluppo di migliori pratiche e strumenti specifici per l’IA aiuta a garantire che i prodotti siano sviluppati, testati e monitorati in modo coerente e affidabile.
Ciò include la creazione di protocolli standardizzati per la valutazione della sicurezza e dell’efficacia dei prodotti IA e la definizione di procedure per il monitoraggio continuo delle loro prestazioni.
4. Sostenere la ricerca relativa alla valutazione e al monitoraggio delle prestazioni dell’IA e anche attraverso la ricerca in rete
Infine, è cruciale sostenere la ricerca continua per valutare e monitorare le prestazioni dell’IA nei prodotti medici.
Questo include la ricerca sulla sicurezza, l’efficacia e l’affidabilità dell’IA, nonché la ricerca collaborativa in rete che coinvolge vari enti e istituzioni.
Tali sforzi di ricerca aiutano a identificare eventuali problemi e a migliorare continuamente le tecnologie IA utilizzate nei prodotti medici. La ricerca in rete, che consente la collaborazione tra diverse organizzazioni e la condivisione di dati e risorse, è fondamentale per avanzare rapidamente in questo campo.
Questo approccio olistico aiuta ad affrontare le prestazioni continue del modello, la gestione del rischio e la conformità normativa dei sistemi AI nelle applicazioni del mondo reale.
È fondamentale che la gestione dell’AI sia basata su un quadro normativo basato sul rischio, costruito su principi robusti, standard, migliori pratiche e strumenti all’avanguardia di scienza normativa.
Questo quadro deve poter essere applicato a tutte le applicazioni AI e adattato allo specifico prodotto medico in questione.
Nella fase di sviluppo, è possibile integrare e arricchire i modelli esistenti con nuovi dati, sviluppando nuovi modelli e stabilendo un monitoraggio della performance per valutarne l’efficacia.Importanti in questa fase sono anche il Risk Management e il Change Management.
L’attuazione di buone pratiche di machine learning (GMLP) durante l’intero ciclo di vita del prodotto è essenziale per promuovere dispositivi medici sicuri, efficaci e di alta qualità basati su AI.
Queste pratiche dovranno evolversi di pari passo con il progresso tecnologico in questo campo in rapida evoluzione.
Collaborazione globale per l’intelligenza artificiale standard

la FDA continua a collaborare attivamente con enti regolatori globali e altre parti interessate.
Per promuovere ulteriormente questa partnership collaborativa, l’Agenzia intende sollecitare input da una gamma di stakeholder su aspetti critici dell’uso dell’AI nei prodotti medici, come la trasparenza, la spiegabilità, la governance, i bias, la sicurezza informatica e l’ assicurazione della qualità.
Questo coinvolgimento delle parti interessate è essenziale per sviluppare un quadro normativo robusto e completo.Inoltre, la FDA mira a promuovere lo sviluppo di iniziative educative per supportare enti regolatori, professionisti sanitari, pazienti, ricercatori e industria mentre navigano nell’uso sicuro e responsabile dell’AI nello sviluppo e nell’applicazione dei prodotti medici.
Queste iniziative possono contribuire a colmare le lacune di conoscenza e garantire che tutte le parti interessate siano adeguatamente informate e preparate.
La collaborazione tra i centri per i prodotti medici della FDA, gli sviluppatori, i gruppi di pazienti, il mondo accademico e altre parti interessate è fondamentale per coltivare un approccio normativo incentrato sul paziente.
Ciò richiede un impegno continuo per la trasparenza, la comunicazione e la condivisione delle conoscenze tra tutte le parti coinvolte.In definitiva, attraverso una stretta collaborazione con i partner globali e il coinvolgimento di un’ampia gamma di stakeholder, la FDA mira a promuovere standard, linee guida e migliori pratiche armonizzate a livello internazionale per l’uso dell’AI nei prodotti medici, garantendo al contempo un approccio normativo incentrato sul paziente che dia priorità alla sicurezza, all’efficacia e all’equità.
Approcci normativi per l’innovazione AI

Questo sforzo include il monitoraggio continuo e la valutazione di tendenze e questioni emergenti per identificare potenziali lacune di conoscenza e opportunità, anche nelle sottomissioni normative.
Ciò consente adattamenti tempestivi che forniscono chiarezza sull’uso dell’AI durante l’intero ciclo di vita del prodotto medico.Inoltre, la FDA sosterrà gli sforzi della scienza normativa per sviluppare metodologie per la valutazione degli algoritmi di AI, identificare e mitigare i bias e garantire la robustezza e la resilienza degli algoritmi di AI per resistere ai cambiamenti negli input clinici e nelle condizioni.
L’agenzia intende anche sfruttare e continuare a costruire sulle iniziative esistenti per la valutazione e la regolamentazione dell’uso dell’AI nei prodotti medici e nello sviluppo dei prodotti medici, inclusa la produzione.
In particolare, la FDA prevede di emettere diverse linee guida riguardanti l’uso dell’AI nello sviluppo e nei prodotti medici stessi, tra cui:
- Linee guida finali sulle raccomandazioni per la sottomissione di marketing per piani di controllo delle modifiche predeterminate per le funzioni software dei dispositivi abilitati all’AI.
- Bozza di linee guida sulle considerazioni per la gestione del ciclo di vita e le raccomandazioni per la sottomissione pre-mercato per le funzioni software dei dispositivi abilitati all’AI.
- Bozza di linee guida sulle considerazioni per l’uso dell’AI a supporto delle decisioni normative per farmaci e prodotti biologici.
Queste linee guida mirano a fornire indicazioni chiare e complete sull’uso dell’AI nello sviluppo dei prodotti per garantire la prevedibilità normativa.
Ciò consentirà alla FDA di adattarsi al mutabile panorama dell’AI e di fare chiarezza sull’utilizzo durante l’intero ciclo di vita del prodotto medico.In sintesi, attraverso il monitoraggio continuo, il sostegno alla scienza normativa e l’emissione di linee guida mirate, la FDA sta lavorando per sviluppare approcci normativi che supportino l’innovazione responsabile e sicura dell’AI nei prodotti medici, garantendo al contempo la protezione della salute pubblica.
Standard e linee guida per l’AI medica
I centri per i prodotti medici della FDA sono impegnati a mantenere elevati standard di sicurezza ed efficacia per i prodotti medici che incorporano tecnologie di intelligenza artificiale (AI)
Basandosi sui principi guida delle Good Machine Learning Practice (GMLP) recentemente pubblicati in collaborazione con Health Canada e MHRA del Regno Unito, l’Agenzia prevede una serie di azioni per promuovere lo sviluppo di standard, linee guida, migliori pratiche e strumenti per l’intero ciclo di vita di questi prodotti innovativi
In primo luogo, la FDA continuerà a perfezionare e sviluppare considerazioni per la valutazione dell’uso sicuro, responsabile ed etico dell’AI nel ciclo di vita dei prodotti medici.
Ciò include fornire un’adeguata trasparenza e affrontare le preoccupazioni relative alla sicurezza e alla cybersecurity di questi sistemi.
Inoltre, l’Agenzia si impegna a identificare e promuovere le migliori pratiche per il monitoraggio a lungo termine della sicurezza e delle prestazioni nel mondo reale dei prodotti medici abilitati all’AI.
Questo è fondamentale per garantire che tali dispositivi mantengano elevati standard di qualità e sicurezza durante l’intero ciclo di vita.
Un altro aspetto chiave è l’esplorazione delle migliori pratiche per documentare e garantire che i dati utilizzati per addestrare e testare i modelli di AI siano idonei all’uso previsto, inclusa un’adeguata rappresentazione della popolazione target.
Ciò è essenziale per limitare i rischi di bias e pregiudizi che potrebbero influire negativamente sulle prestazioni e sull’equità di questi sistemi.Infine, la FDA intende sviluppare un quadro e una strategia per l’assicurazione della qualità degli strumenti o sistemi abilitati all’AI utilizzati nel ciclo di vita dei prodotti medici, con un’enfasi sul monitoraggio continuo e la mitigazione dei rischi.
In conclusione, attraverso queste iniziative, i centri per i prodotti medici della FDA mirano a promuovere lo sviluppo di standard, linee guida, migliori pratiche e strumenti robusti per supportare l’uso sicuro, efficace e responsabile dell’AI nei prodotti medici durante l’uso intero ciclo di vita.
Ciò richiederà una stretta collaborazione con tutte le parti interessate e un impegno continuo per l’innovazione e la protezione della salute pubblica in questo campo in rapida evoluzione.
Progetti dimostrativi per valutare l’AI

La FDA intende supportare progetti che considerino le disuguaglianze sanitarie associate all’uso dell’AI nello sviluppo dei prodotti medici.
L’obiettivo è promuovere l’equità e garantire la rappresentatività dei dati utilizzati per addestrare e testare i modelli di AI, sfruttando gli sforzi in corso per la diversità, l’equità e l’inclusione.
Ciò è essenziale per evitare che i bias e le disuguaglianze esistenti vengano amplificati o perpetuati dai sistemi di AI.
Un altro aspetto chiave è il supporto al monitoraggio continuo delle prestazioni e dell’affidabilità degli strumenti di IA utilizzati nello sviluppo dei prodotti medici all’interno dei progetti dimostrativi.
Ciò garantirà l’adesione agli standard e il mantenimento delle prestazioni durante l’intero ciclo di vita di questi sistemi.
Questi progetti dimostrativi forniranno informazioni preziose sull’impatto dell’AI sulla sicurezza e l’efficacia dei prodotti medici, consentendo alla FDA di sviluppare politiche e linee guida basate sull’evidenza per regolamentare l’uso di queste tecnologie innovative.In sintesi, attraverso il supporto a progetti mirati che affrontano domande critiche come il bias, le disuguaglianze sanitarie e il monitoraggio continuo delle prestazioni, la FDA mira a promuovere l’uso sicuro, equo ed efficace dell’AI nello sviluppo dei prodotti medici.
Ciò richiederà una stretta collaborazione con ricercatori, sviluppatori e altre parti interessate per garantire che questi progetti forniscano informazioni preziose e attuabili per guidare il processo decisionale normativo in questo campo in rapida evoluzione.